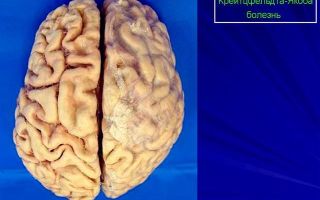
Что такое болезнь Крейтцфельдта-Якоба?
Болезнь Крейтцфельдта-Якоба (болезнь Кройцфельдта-Якоба, БКЯ) — это редкое неврологическое заболевание (заболевание нервной системы), которое вызывает повреждение головного мозга.
Он относится к группе заболеваний, называемых трансмиссивными губчатыми энцефалопатиями, или прионными заболеваниями, которые поражают людей и животных.
БКЯ смертельна, а лечения никакого нет, если только симптоматическое. Возникает болезнь из-за неправильного белка, называемого прионом, загрязняющего нервную систему.
Прионы
Прионы — это инфекционные частицы, состоящие из ненормально сложенного белка. В некоторых отношениях прион похож на вирус, так как он может размножаться и вызывать заболевание. Но, в отличие от вируса, он сделан полностью из белка и не имеет генетического материала.
Это делает прионы намного жестче, чем вирусы или бактерии. Они могут выдерживать экстремальные температуры и радиацию, и устойчивы к расщеплению ферментами, обычно контролирующими уровень белка в организме. Антибиотики и противовирусные препараты на них не влияют.
Прионы убивают клетки головного мозга и создают в мозге отверстия, придавая губчатый вид.
Типы болезни Крейтцфельдта-Якоба
Существует 4 различных типа болезни Крейтцфельдта-Якоба, каждый из которых имеет свою причину:
- Спорадическая (sCJD) является наиболее распространенной формой, на которую приходится 85% случаев болезни. Причина неизвестна, но обычно она затрагивает людей старше 40 лет.
- Новый вариант (nvCJD) был впервые идентифицирован в 1996 году. nvCJD вызывается употреблением мяса крупного рогатого скота, инфицированного коровьим бешенством. nvCJD в основном поражает людей в возрасте от 20 лет.
- Ятрогенная форма (1CJD) — инфекция передается от человека с заболеванием посредством медицинского или хирургического лечения. В наши дни его случаи крайне редки.
- Наследственная форма (fCJD) — это редкая форма прионной болезни, вызванная наследованием дефектного гена, продуцирующим прионы.
Симптомы и стадии
Симптомы и течение болезни Крейтцфельдта-Якоба различаются в зависимости от типа. Тем не менее, все четыре типа болезни имеют некоторые схожие признаки, описанные ниже.
Ранние стадии
- Болезнь Крейтцфельдта-Якоба обычно начинается с эмоциональных или поведенческих проблем, таких как депрессия, беспокойство или возбуждение.
- Бред (сильные убеждения в вещах, явно не соответствующих действительности) и галлюцинации (видение или слышание вещей, которых нет) типичны для нового варианта болезни (nvCJD).
- У пациентов также развиваются неврологические проблемы (влияющие на нервную систему), такие как боль или онемение в частях тела.
- В течение нескольких недель человек быстро ухудшается, становится запутанным и испытывает потерю памяти (симптомы, типичные для слабоумия).
Более поздние стадии
Наряду с растерянностью и потерей памяти, человек может потерять координацию и равновесие, и начать слепнуть.
Месяцы спустя больные не могут ходить, разговаривать и заботиться о себе. Они не знают своего окружения и у них развиваются резкие движения мышцами.
Три четверти людей со спорадической (классической) формой болезнью Крейтцфельдта-Якоба умирают в течение шести месяцев после постановки диагноза, часто от инфекции пневмонии. Другие умирают в течение нескольких недель.
Причины болезни Крейтцфельдта-Якоба
Болезнь вызывается инфекционным белком в мозге, называемым прионом.
Роль белков в мозге
Белки — это молекулы, состоящие из аминокислот, помогающие клеткам нашего организма функционировать.
Белки начинаются как цепочка аминокислот, которые затем складываются в трехмерную форму. Такое “складывание белка” (фолдинг белка) позволяет им выполнять полезные функции в наших клетках.
Прионные белки (которые не являются теми же самыми инфекционными прионами, которые вызывают БЯК, т.е. нормальные прионные белки) — это тип белков, встречающихся в мозгу и некоторых других тканях нервной системы. Точная роль прионных белков в мозгу неизвестна, но считается, что они могут иметь какое-то отношение к долгосрочной памяти.
Когда складывание белка идет не правильно
Иногда ошибки происходят во время фолдинга белка, в результате прионный белок не может быть использован организмом. Эти неправильно уложенные прионные белки обычно перерабатываются организмом, но иногда они могут накапливаться, что может вызвать проблемы, такие как болезнь Альцгеймера.
Прионы — это неправильно свернутые прионные белки, которые проникают в клетки мозга и также приводят к неправильному складыванию нормальных белков. Это приводит клетки мозга к смерти, высвобождая больше прионов, заражая другие клетки головного мозга.
В конце концов, кластеры клеток головного мозга погибают и заменяются отложениями прионов, называемыми бляшками. Эти бляшки производят небольшие отверстия в мозге, придавая ему губчатый вид. Повреждение головного мозга вызывает умственные и физические нарушения и возможную смерть, связанную с болезнью.
Прионы могут жить в нервной ткани (в головном или спинном мозге), в течение очень долгого времени, даже после смерти человека или животного.
Спорадическая (классическая) форма
Спорадическая БЯК (sCJD) является наиболее распространенным типом болезни, хотя все еще очень редко. Неизвестно, что вызывает sCJD, но она может быть связано с тем, что нормальный белок самопроизвольно превращается в прион или нормальный ген самопроизвольно превращается в дефектный ген, продуцирующий прионы. sCJD обычно поражает людей старше 40 лет.
Новый вариант
Существуют явные доказательства того, что новый вариант БЯК (vCJD) вызван тем же штаммом прионов, что и губчатая энцефалопатия крупного рогатого скота (также известная как коровье бешенство).
Исследование правительства Великобритании в 2000 году пришло к выводу, что прион распространялся через крупный рогатый скот, которого кормили мясокостной смесью, содержащей следы зараженного мозга или спинного мозга. Затем прион оказался в переработанных мясных продуктах, таких как бифбургеры, и вошел в пищевую цепь человека.
Ятрогенная форма
Ятрогенная форма болезни является результатом передачи инфекции от человека с БЯК посредством медицинского или хирургического лечения.
Большинство ятрогенных случаев болезни Крейтцфельдта-Якоба (1CJD) произошло при использовании гормона роста человека, который использовался для лечения детей с ограниченным ростом.
Между 1958 и 1985 годами тысячи детей лечились гормоном, который в то время был извлечен из гипофиза (железа, находящегося у основания черепа) человеческих трупов.
У крошечного меньшинства этих детей развился БЯК, поскольку полученные ими гормоны были взяты из желез, инфицированных людей.
Несколько других случаев 1CJD произошли, когда люди получили пересадку зараженной ткани или вступили в контакт с хирургическими инструментами, которые были заражены болезнью Крейтцфельдта-Якоба. Это произошло из-за того, что прионы жестче, чем вирусы или бактерии, поэтому нормальный процесс стерилизации хирургических инструментов не работал.
Наследственная форма заболевания
Эта очень редкая форма болезни, вызвана она наследственной мутацией (дефектом) гена, который производит нормальные белки. Похоже, что измененный ген продуцирует прионы, вызывающие БЯК.
У каждого есть две копии своих генов, но мутированный ген является доминирующим. Это означает, что человеку нужно унаследовать только один мутированный ген для развития болезни.
Может ли болезнь Крейтцфельдта-Якоба передаваться от человека к человеку?
Теоретически, болезнь может передаваться от больного человека другим, но только через инъекцию или потреблении мозговой ткани.
Считается, что болезнь Крейтцфельдта-Якоба не передается при обычном повседневном контакте с больными или воздушно-капельным путем, кровяным контактом или половым контактом.
Диагностика
Диагноз обычно основывается на истории болезни, симптомах и серии анализов (см. ниже). Невролог (врач, специализирующийся на состояниях нервной системы) проведет тесты, чтобы исключить другие состояния с похожими симптомами, такие как болезнь Альцгеймера, болезнь Паркинсона или опухоль головного мозга.
Единственный способ подтвердить диагноз болезни Крейтцфельдта-Якоба — это непосредственно исследовать ткань головного мозга с помощью биопсии головного мозга (см. ниже) или, если человек умер, после вскрытия.
Проверка на общие признаки
Клинический невролог исключит другие заболевания с похожими симптомами и проверит наличие некоторых общих признаков болезни, выполнив любой из следующих тестов:
- Магнитно-резонансная томография (МРТ) головного мозга. При ней используются сильные магнитные поля и радиоволны для получения детального изображения мозга, и могут обнаруживаться отклонения, характерные для БЯК.
- Электроэнцефалограмма (ЭЭГ). Исследование регистрирует активность мозга и может обнаруживать аномальные электрические паттерны, наблюдаемые при спорадической форме БЯК.
- Люмбальная пункция. В нижнюю часть позвоночника вводится игла для отбора пробы спинномозговой жидкости (которая окружает головной и спинной мозг) для исследования в лаборатории. Если в жидкости обнаруживается белок, называемый 14-3-3, это указывает на то, что возможно человек болен болезнью Крейтцфельдта-Якоба (этот белок обнаруживается почти во всех случаях классической формы заболевания и в 50% случаев нового варианта болезни).
- Биопсия миндалин. Небольшой кусочек ткани берется из миндалин и проверяется на наличие аномальных прионов, обнаруживаемых при новом варианте БЯК (их нет в других типах).
- Генетический тест. Это простой анализ крови, показывающая есть ли мутация (ошибка) в гене, который производит нормальный белок. Положительный результат может указывать на наследственную форму заболевания.
Во время биопсии головного мозга хирург просверливает крошечное отверстие в черепе и удаляет маленький кусочек мозговой ткани с помощью очень тонкой иглы. Это делается под общим наркозом (человек спит).
Поскольку биопсия головного мозга несет в себе риск вызвать повреждение головного мозга или судороги (эпилепсию), она проводится только в нескольких случаях, когда существует опасение, что у пациента нет болезни Крейтцфельдта-Якоба, но есть какое-то другое излечимое состояние.
Лечение
- Не существует доказанной терапии или лечения какой-либо формы болезни Крейтцфельдта-Якоба.
- Множество потенциальных терапевтических вмешательств при болезни остаются в настоящее время на уровне дискуссии.
- Лечение включает попытки сохранить человеку максимально комфортно короткую жизнь и уменьшить симптомы с помощью лекарств.
Например:
- Психологические симптомы, такие как беспокойство и депрессия, можно лечить с помощью седативных средств и антидепрессантов.
- Другие лекарства, такие как клоназепам и вальпроат натрия, могут быть использованы для лечения мышц и тремора.
- Обезболивающие препараты на основе опиатов могут обеспечить эффективное обезболивание.
Команда специалистов
Когда кому-то ставят диагноз БЯК, его направляют в соответствующий центр для диагностики и лечения. Врач и медсестра из этих служб будут назначены пациенту для связи с местными службами, такими как врач общей практики, социальный работник, физиотерапевт и специалист по трудотерапии.
Доступны команды специалистов для диагностики и оказания клинической и эмоциональной поддержки пациентам и их семьям, а также работа вместе с местной командой по уходу. В местную команду по уходу могут входить врачи и медсестры, специалисты по гигиене труда, диетологи, консультанты по вопросам недержания и социальные работники.
Уход и поддержка
По мере прогрессирования заболевания людям с болезнью Крейтцфельдта-Якоба потребуется значительный уход и практическая поддержка.
Помимо помощи в кормлении, мытье и передвижении, некоторые люди могут нуждаться в помощи при мочеиспускании. Часто требуется использование катетера (трубки, которая вставляется в мочевой пузырь и используется для слива мочи).
У многих людей возникают проблемы с глотанием, поэтому им, возможно, придется питаться и пить через трубку для кормления.
Может быть возможно лечить людей с БЯК и в домашних условиях, но это будет зависеть от прогрессирования и тяжести заболевания.
Давление при заботе о ком-то с БЯК может быть трудным. Вместо того, чтобы справляться с проблемой в домашних условиях, многие лица, осуществляющие уход, предпочитают пользоваться услугами больниц.
Прогноз
Не существует лекарство от БКЯ. Болезнь быстро ухудшает состояние человека до такой степени, что он больше не может заботиться о себе и не может двигаться или говорить.
Большинство людей со спорадической формой БКЯ умирают в течение 6 месяцев после постановки диагноза, часто от пневмонии. Люди с новым вариантом болезни живут в среднем чуть больше года.
Профилактика
Поскольку связь между коровьем бешенством и болезнью Крейтцфельдта-Якоба была подтверждена, предупредить развитие заболевания поможет качественная термическая обработка мяса животных, а также повышение иммунной системы организма.
Источник: https://tvojajbolit.ru/nevrologiya/bolezn_kreyttsfeldta_yakoba/
Болезнь Крейтцфельдта-Якоба
Болезнь Крейтцфельдта-Якоба — редко встречающееся дегенеративное заболевание головного мозга, связанное с накоплением в нейронах патологического белка приона. Клинически болезнь Крейтцфельдта-Якоба проявляется слабоумием, пирамидными и экстрапирамидными нарушениями, миоклониями, симптомами поражения мозжечка и нарушением зрения. Диагноз болезни Крейтцфельдта-Якоба основывается на совокупности клинических симптомов, данных ЭЭГ, анализа цереброспинальной жидкости, МРТ и ПЭТ головного мозга, а также морфологического исследования образца тканей мозга, полученного в результате биопсии или посмертно. Эффективное лечение болезни Крейтцфельдта-Якоба пока не найдено. Заболевание имеет 100% летальный исход.
Болезнь Крейтцфельдта-Якоба очень редкое заболевание. Ранее в литературе указывалось, что частота его встречаемости примерно 1 случай на 1 млн. человек населения.
Однако в 90-х годах прошлого века начали отмечаться случаи так называемого нового варианта болезни Крейтцфельдта-Якоба, связанного с заражением от крупного рогатого скота и названного «коровьим бешенством». Только в Англии за 5 лет от этого заболевания умерло 86 человек.
Наиболее распространена болезнь Крейтцфельдта-Якоба среди людей в возрасте 65-70 лет и старше. Однако в новом варианте болезнь Крейтцфельдта-Якоба зачастую поражает лиц молодого возраста. Относительно высокий уровень заболеваемости отмечается в Англии, Израиле, Чили и Словакии.
Болезнь Крейтцфельдта-Якоба
Установлено, что болезнь Крейтцфельдта-Якоба имеет инфекционный характер.
Заражение может произойти при пересадке зараженных прионами тканей, через нейрохирургический инструмент и препараты крови, при введении некоторых гормональных препаратов (человеческого гонадотропина для лечения бесплодия и соматотропина для терапии гипопитуитаризма). Болезнь Крейтцфельдта-Якоба новой формы может развиваться после употребления в пищу мяса заболевших животных (коровы) или носителей инфекции (овец и коз).
В результате ряда исследований стало известно, что болезнь Крейтцфельдта-Якоба связана с проникновением в организм инфекционного белка — приона. В норме в клетках головного мозга человека содержится здоровый прион, имеющий несколько другое строение.
Инфекционный прион, попадая в организм человека не разрушается, а с током крови поступает в головной мозг и откладывается на поверхности нейронов.
Его взаимодействие с нормальными прионами мозговой клетки приводит к тому, что они изменяют свою структуру, постепенно трансформируясь в патогенную, подобную инфекционному приону, форму. Патогенные прионы образуют бляшки и приводят к гибели нейрона.
Болезнь Крейтцфельдта-Якоба имеет достаточно длительный инкубационный период, связанный с временем, необходимым для проникновения инфекционных прионов в мозговую ткань и патогенной трансформации здоровых прионов.
Длительность инкубационного периода напрямую зависит от способа заражения. При инфицировании тканей головного мозга зараженным хирургическим инструментом болезнь Крейтцфельдта-Якоба развивается через 15-20 месяцев.
При инфицировании через имплантированные в околомозговые структуры ткани (например, твердую мозговую оболочку, роговицу глаза) инкубационный период может длиться до 5,5 лет.
При внутримышечном введении инфицированных лекарственных препаратов (например, гонадотропина, соматотропина, содержащих бычий тромбин гемостатиков) болезнь Крейтцфельдта-Якоба начинает проявляться спустя 12,5 лет.
Отмечаются также наследственные формы болезни Крейтцфельдта-Якоба, связанные с генетическими нарушениями, приводящими к образованию патологических прионов.
Практическая неврология классифицирует болезнь Крейтцфельдта-Якоба с учетом ее клинической формы.
В соответствии с этим выделяют: подострую спонгиоформную энцефалопатию, отличающуюся быстрым течением и диффузным поражением мозговой коры; классическую (дискинетическую) форму, представляющую собой сочетание пирамидных и экстрапирамидных симптомов со слабоумием (деменцией); промежуточную форму болезни Крейтцфельдта — Якоба, характеризующуюся преобладанием мозжечковых и подкорковых расстройств; амиотрофическую форму, двигательные и речевые расстройства при которой напоминают клинику бокового амиотрофического склероза.
Различают также наблюдавшуюся ранее спорадическую форму заболевания и новый вариант болезни Крейтцфельдта-Якоба («коровье бешенство»).
В большинстве случаев болезнь Крейтцфельдта-Якоба характеризуется постепенным развитием, однако возможно подострое или острое начало.
Примерно в 30% случаев болезнь Крейтцфельдта-Якоба начинается с продромальных симптомов: раздражительности, рассеянности, головных болей, нарушений сна, головокружения, ухудшения памяти, снижения зрения, безынициативности, снижения либидо, изменения поведенческих реакций.
Возможно эпизодическое возникновение эйфории и/или беспричинного страха, отрывистые бредовые или галлюцинаторные переживания.
Из неврологических нарушений в продромальном периоде наблюдаются: шаткость во время ходьбы, парестезии, расстройство высших функций коры головного мозга (алексия, акалькулия и пр.). Описаны несколько случаев, когда болезнь Крейтцфельдта-Якоба дебютировала с появления корковой слепоты.
В стадии развернутых клинических проявлений болезнь Крейтцфельдта-Якоба характеризуется прогрессирующим спастическим параличем (параплегией или гемиплегией), атаксией, эпилептическими припадками. Возникают экстрапирамидные нарушения: мышечная ригидность, атетоз, тремор.
Практически у всех больных наблюдаются миоклонии — быстрые неритмичные сокращения отдельных мышц. Чаще всего отмечается миоклонус губы и века. Наблюдаются вторично генерализованные миоклонические приступы.
Появляется и нарастает ярко выраженная деменция, сопровождающаяся нарушениями речи вплоть до ее полного распада. Новый вариант болезни Крейтцфельдта-Якоба отличается преобладанием психиатрической симптоматики и расстройств чувствительности.
В 100% случаев он сопровождается мозжечковыми нарушениями, в то время как при спорадической болезни Крейтцфельдта-Якоба расстройства функции мозжечка наблюдаются лишь в 40% случаев.
В терминальной стадии болезнь Крейтцфельдта-Якоба характеризуется глубокой деменцией. Пациенты не контактны, находятся в состоянии прострации, утрачен контроль над функцией тазовых органов.
Наблюдаются гиперкинезы, выраженные мышечные атрофии, нарушения глотания, пролежни. Возможна гипертермия и эпилептические приступы.
Смерть наступает в коматозном состоянии на фоне децеребрационной ригидности и выраженной кахексии.
Клиническая диагностика заболевания основана на сочетании прогрессирующей в течение 2-х лет деменции, пирамидных и экстрапирамидных расстройств, миоклоний, мозжечковых расстройств и нарушений зрения.
Для уточнения диагноза невролог назначает инструментальные методы обследования: электроэнцефалографию (ЭЭГ), ПЭТ и МРТ головного мозга, люмбальную пункцию.
В сомнительных случаях для установления диагноза болезнь Крейтцфельдта-Якоба производят стереотаксическую биопсию головного мозга.
На ЭЭГ на фоне сниженной биоэлектрической активности у большинства больных наблюдаются периодические или псевдопериодические острые волны.
Отмечается билатеральная, фокальная или генерализованная миоклоническая активность, которая в начальной стадии определяется у половины больных, а в терминальной стадии выявляется в 100% случаев спорадической болезни Крейтцфельдта-Якоба. Новый вариант заболевания часто протекает без существенных изменений ЭЭГ-паттерна.
При проведении МРТ головного мозга в Т2-режиме определяется так называемый «симптом медовых сот» – участки повышенного сигнала, исходящие от подкорковых ганглиев и таламуса.
Зачастую выявляются признаки атрофических изменений мозжечка и коры головного мозга, расширение желудочков и боковых цистерн мозга.
ПЭТ диагностирует зоны пониженного метаболизма, локализующиеся в подкорковых ядрах, полушариях мозжечка и коре мозга.
Люмбальная пункция в обязательном порядке проводится пациентам с подозрением на болезнь Крейтцфельдта-Якоба. Она позволяет оценить давление ликвора и произвести исследование цереброспинальной жидкости. Отсутствие патологических изменений ликвора позволяет дифференцировать болезнь Крейтцфельдта-Якоба от многих других заболеваний ЦНС.
Наиболее достоверным методом диагностики является морфологическое исследование образцов мозговой ткани, которые могут быть получены прижизненно путем биопсии или при аутопсии после смерти пациента. Применение иммуноцитохимического метода позволяет обнаружить в исследуемом материале отложения патологического белка — приона.
Дифференциальную диагностику болезни Крейтцфельдта-Якоба необходимо проводить с лобно-височной деменцией, герпевирусным энцефалитом, болезнью Альцгеймера, мультиинфарктной деменцией (слабоумием, развивающимся после повторных ишемических и геморрагических инсультов), хроническим менингитом, арахноидитом, нормотензивной гидроцефалией, энцефалопатией Хашимото, сопровождающей некоторые случаи аутоиммунного тиреоидита, и др.
В современной медицине подходы к лечению болезни Крейтцфельдта-Якоба находятся в стадии активной разработки. Общепринятые противовирусные препараты, а также пассивная иммунизация и вакцинация людей и животных выявились неэффективными.
Отмечено, что блокирующее действие на синтез патологических прионов в инфицированных нейронах оказывает Брефелдин А, а блокаторы кальциевых каналов продлевают жизнь инфицированных клеток. Обычно пациенты, имеющие болезнь Крейтцфельдта-Якоба, получают симптоматическое лечение.
Оно направлено на купирование миоклонических приступов и экстрапирамидных нарушений, в связи с чем применяются антиэпилептические и противопаркинсонические лекарственные средства.
Болезнь Крейтцфельдта-Якоба является фатальным заболеванием. Продолжительность жизни большинства больных не превышает 1 год с момента начала клинических проявлений; а средняя длительность составляет 8 месяцев. Лишь 5-10% заболевших живут в течение 2 и более лет. Наследственная болезнь Крейтцфельдта-Якоба в среднем длится около 26 месяцев.
Источник: https://www.KrasotaiMedicina.ru/diseases/zabolevanija_neurology/creutzfeldt-jakob
Болезнь Крейцфельдта-Якоба
Болезнь Крейтсфельдта-Якоба (БКЯ) — это прогрессирующее заболевание головного мозга, спинного мозга и базальных ганглиев. Считается основным видом прионной болезни, также известной как губчатая энцефалопатия.
Синдром Крейцфельдта-Якоба составляет 85% от всех прионных заболеваний человека.
Если говорить простыми словами, то прионные белки (чуждые организму человека, попадающие извне) в организме проникают в нервную клетку и модифицируют нормальный белок, превращая его в себе подобный.
Этот белок клетка не может удалить или уничтожить и погибает сама. Процесс необратим и развивается в геометрической прогрессии.
БКЯ была описана в 1920 году Г.Г. Крейтцфельдтом, а годом позже А. Якоб указал, что эта патология мозга сочетается с нарушениями психики и симптомами повреждения спинного мозга. Им же были обнаружены признаки поражения пирамидной и экстрапирамидной систем, отвечающих за координацию движений (пирамидная) и за поддержание тонуса мышц и поз (экстрапирамидная).
Причины болезни Крейтцфельдта-Якоба
Причина губчатой энцефалопатии — накопление мутировавшего прионного белка в нейронах. Сам механизм довольно сложный. У белка есть первичная, вторичная и третичная структуры. Первичная — это последовательность аминокислот, вторичная — структура, образованная водородными связями (белок более компактный).
Когда происходит ошибка в экспрессии гена (процесс, в ходе которого информация с гена «считывается», и мы имеем готовый продукт — белок или РНК), развивается спонтанная или наследственная БКЯ. Если произошло заражение, то инициатором смены третичной структуры является привнесенный белок.
Вторичная структура белка — это спирали, образованные водородными связями. При мутации белки не скручиваются в альфа-спирали, а складываются в бета-лист — инфекционное состояние. И такие белки способны видоизменять нормальные спирали. Бета-листы объединяются в плоские волокна, которые образуют поры в тканях мозга.
Это и есть прионная болезнь Крейтцфельдта-Якоба.
Пути передачи болезни Крейтцфельдта-Якоба
Самый распространенный путь передачи энцефалопатии — попадание белка-мутанта и накопление его в нейронах с одновременным нарушением функций ткани и со временем полной ее деградации.
Источник модифицированного прионного белка — зараженное мясо, поэтому у заболевания Крейтцфельдта-Якоба есть синоним — губчатый (или губкообразный) энцефалит крупного рогатого скота, или коровье бешенство (не путать с вирусным).
В истории были случаи, когда больные заражались при пересадке тканей или из-за нестерильных инструментов, применяемых при операциях на открытом мозге.
Чтобы уменьшить опасность распространения заболевания от животных, ЕС запретил использовать ткани с подозрением на заражение, а с 2001 года забитые животные исследуются на наличие возбудителя БКЯ.
Еще один путь передачи болезни Крейтцфельдта-Якоба — по наследству. Ребенку передается ген, продуктом экспрессии которого является тот самый прионный белок-мутант. Чаще всего такие мутации возникают спонтанно, эпизодов, когда болезнь переходила от одного поколения к другому, очень мало.
Хотя в данном случае причина может быть в том, что некоторые люди просто не доживают до манифеста заболевания, которое начинает проявляться примерно в 40–50 лет, и ученые пока не выяснили, почему так долго ген «молчит».
Самое страшное — из-за позднего дебюта болезни люди уже в основном имеют детей, и принимать профилактические меры можно только в отношении внуков. Вероятность передачи заболевания составляет 50%.
При наследственной БКЯ белок экспрессируется («воспроизводится») геном неверно. Это приводит к тому, что в нейронах накапливаются прионные палочки, что и нарушает функции мозга. Название «губчатый энцефалит» неслучайно — мозг больных буквально похож на пористую губку.
Это заболевание неизлечимо, и срок жизни пациента очень короткий, обычно 2–3 года. В редких случаях болезнь прогрессирует быстро, и человек умирает буквально за несколько месяцев после дебюта заболевания. Среди всех известных случаев БКЯ унаследованные составляют не более 10%.
Есть и спорадические формы болезни, при которых она появляется без явной причины, обычно это классический тип. На такие заболевания приходится порядка 85% всех случаев.
Факторы риска болезни Крейтцфельдта-Якоба
В большинстве случаев причины заболевания определить невозможно, но все же есть факторы риска:
- возраст (классическая форма манифестирует обычно после 60 лет, семейная — раньше, вариантная БКЯ — примерно в 20–30 лет);
- генетика (при наличии заболевания у одного из родителей шансы передачи его ребенку любого пола составляют 50%);
- воздействие зараженного материала.
В последнем случае речь не о мясе и частях туши, а скорее о трансплантациях тканей, хотя и употребление внутрь (с едой) также может привести к заражению коровьим бешенством.
Симптомы болезни Крейтцфельдта-Якоба
Клинические проявления БКЯ:
- слабоумие — прогрессирует очень быстро, часто его называют опустошающей деменцией, так как в очень короткие сроки происходит полный распад личности, человек буквально увядает;
- нарушаются все корковые функции, угасают пирамидные и экстрапирамидные функции (движения становятся несогласованными, сложно удерживать позу);
- миоклонус (кратковременные спонтанные сокращения мышц);
- нарушения речи;
- эпилептические припадки;
- разнообразные нарушения зрения (чаще всего — диплопия (двоение в глазах) и галлюцинации).
Продромальный период
На начальном этапе болезни Крейтцфельдта-Якоба симптомы возникают приблизительно у трети больных за несколько недель или месяцев до проявления деменции:
- нарушение сна;
- снижение памяти, остроты мышления;
- меняется поведение;
- астения;
- происходит потеря веса из-за ухудшения аппетита.
Инициальный период
Этот период характеризуется разнообразными нарушениями зрения (изображение двоится или становится мутным), головными болями, человек теряет чувство равновесия, у него кружится голова, присутствует общая слабость и апатия. Развитие болезни постепенное, реже — острое.
Развернутый период
Все описанные выше симптомы Кретцфельдта-Якоба «раскрываются» в полной мере — двигательные нарушения, парезы, тремор, ригидность, ухудшение зрения, мышечная фибрилляция.
Если развивается спорадическая форма болезни, то она прогрессирует очень быстро, результат — опустошающая деменция. Средняя продолжительность жизни после первых симптомов — 8 месяцев. При подостром течении смерть может наступить через 1–2 года после манифестации БКЯ. Наследственная форма дает пациенту примерно 24–26 месяцев.
Болезнь Крейтцфельдта-Якоба представлена в нескольких формах:
- Спонтанная (она же классическая, спорадическая), при которой прионы появляются в мозге спонтанно, в основном у пожилых людей с очень низким риском (1 случай на 1 000 000 человек). Болезнь манифестирует потерями памяти (в основном короткими), резкой сменой настроения (лабильность), апатией. Конечная фаза этой формы — нарушения зрения, галлюцинации разного рода, расстройства речи. Течение у спорадической формы обычно подострое, у 40% больных нарушены когнитивные функции (память, работоспособность и т.д.), у 40% — мозжечковые нарушения, у 20% комбинированы когнитивные и мозжечковые нарушения. Терминальная стадия болезни Крейтцфельдта-Якоба характеризуется серьезными когнитивными нарушениями, распадом личности и полной апатией.
- Ятрогенная форма — это развитие заболевания вследствие случайного внесения мутантных белков-прионов во время медицинского вмешательства. То есть в данном случае причина болезни Крейтцфельдта-Якоба — заражение. Течение заболевания схоже с классической (спорадической, спонтанной) формой.
- ВБКЯ (вариантная БКЯ) приобретенная — диагностировалась после приема мяса и частей туши зараженного животного. Эта форма дебютирует в молодом возрасте, больные теряют интерес к увлечениям, страдают от депрессий. Главное отличие от классической формы — пациент осознает, что ему становится хуже.
Диагностика болезни Крейтцфельдта-Якоба
Деменция при болезни Крейтцфельдта-Якоба развивается в 100% случаев. Поэтому если имеет место быстро прогрессирующее слабоумие, всегда предполагают БКЯ.
Она считается определенной, если: выявлены скрепи-ассоциированные фибриллы (особым образом полимеризованные белки), характерные неврологические, патолого-анатомические и морфологические симптомы, а также обнаружен протеазо-резистентный PrP.
БКЯ вероятна, если у пациента быстро прогрессирует слабоумие, есть нетипичные изменения на электроэнцефалограмме и как минимум два признака из списка:
- миоклонус;
- снижение зрения;
- нарушения согласованности движения (пирамидные и экстрапирамидные);
- симптомы повреждения мозжечка;
- акинетический мутизм (состояние пациента, когда он теряет способность двигаться и говорить, но физически способен это делать).
Инструментальные методы для диагностики болезни Крейтцфельдта-Якоба — МРТ, позитронно-эмиссионная томография, ЭЭГ, тесты для когнитивных функций, анализ ликвора, биопсия и посмертная гистология и морфология тканей.
Необходима дифференциальная диагностика с таким заболеваниями, как васкулит нервной системы; болезнь Альцгеймера (быстро прогрессирующая); энцефалопатия Хашимото (аутоиммунная с высоким уровнем тиреодных тел, реагирует на кортикостероиды); внутрисосудистая лимфома; деменция с белковыми телами (Леви); отравление литием или висмутом.
Лечение болезни Крейтцфельдта-Якоба
Саму болезнь остановить нельзя, но можно облегчить ее течение. Противовирусные препараты оказались неэффективными (так как возбудитель — не вирус Крейтцфельдта-Якоба, а просто белок), а многие возможные терапевтические вмешательства остаются в настоящее время на уровне обсуждения. Вакцинация также не работает.
Для лечения сейчас применяют:
- препарат для разрушения аппарата Гольджи;
- препараты для блокирования кальциевых каналов — увеличивают срок жизни инфицированных нейронов;
- синтетический иммуномодулятор, который при длительном применении вызывает увеличение количества гликозамингликанов (мукополисахаридов) в нейронах и замедляет накопление мутировавших белков, но он может ухудшать состояние мембран из-за нарушения фосфолипидного обмена.
Симптоматическое лечение Крейтцфельдта-Якоба купирует миоклонические приступы и экстрапирамидные нарушения — для этого используют те же препараты, что и при лечении эпилепсии и болезни Паркинсона.
Осложнения болезни Крейтцфельдта-Якоба
Учитывая, что болезнь развивается довольно быстро и, по сути, является просто уничтожением нейронов, трудно говорить об осложнениях, ведь пациент все равно умирает. Лечением можно только отсрочить смерть и облегчить симптомы.
Профилактика болезни Крейтцфельдта-Якоба
Это заболевание смертельно, эффективного лечения нет, поэтому профилактика болезни Крейцтфельдта-Якоба очень важна.
Специалистам, которые могут контактировать с зараженными тканями и/или биологическими жидкостями, нужно работать только в перчатках и не допускать попадания материала на слизистые. Если зараженный материал попадает на кожу человека, то сначала нужно провести дезинфекцию 4-процентным раствором щелочи в течение 10 минут, а затем тщательно промыть проточной водой.
Материалы, которые контактировали с зараженными жидкостями или тканями, а также с тканями пациентов с возможной БКЯ, подвергаются паровому автоклавированию при температуре 132 градуса (1 час) или погружению в 1h раствор щелочи или 10-процентный раствор гипохлорита натрия. Стандартные методы стерилизации в случае губчатой энцефалопатии неэффективны.
Профилактика ятрогенной болезни Крейтцфельдта-Якоба
Ятрогенная форма — это тип БКЯ, при котором прионы вносятся в тело человека при медицинском вмешательстве. Источником белков-мутантов в данном случае служат нестерильные инструменты и зараженные образцы ткани от мертвых пациентов. Согласно данным, сейчас этот источник заражения устранен.
Профилактика коровьего бешенства
Болезнь Крейтцфельдта-Якоба, или коровье бешенство, имеет очень интересную историю. В 1980-х годах Англию потрясла эпидемия смертельной болезни крупного рогатого скота. Симптомы были очень схожи с бешенством, но механизм возникновения отличался. При исследовании оказалось, что это губчатая энцефалопатия.
Название прикрепилось потому, что мозг умершего животного напоминал губку. От этого вида энцефалопатии страдают не только коровы, но и овцы, козы, грызуны и даже домашние кошки. У людей это болезнь куру и БКЯ (причина первой — в поедании мозга умерших людей). Выяснили, что причина столь быстрого распространения «нового бешенства» — мясокостная мука.
Сменили технологию ее изготовления, и возбудитель сохранялся в продукте.
С эпидемией в Европе благополучно справились, но еще в 1996 году в РФ прекратили импорт мяса и мясопродуктов из стран Западной Европы.
Если ввозят мясопродукты, например, на корм домашним питомцам, то производителем должно быть гарантировано отсутствие в составе европейского мяса. Также запрещен ввоз племенного скота.
Однако не стоит отказываться от самого мяса — оно не заражено, ведь прионы «оседают» только в нейронах.
Сейчас с определенной долей уверенности можно говорить, что губчатая энцефалопатия побеждена. Но нужно учитывать, что диагностика возможна только посмертно, а инкубационный период заболевания очень большой.
Источник: https://yusupovs.com/bolezn-kreytsfeldta-yakoba/
Болезнь Крейтцфельдта-Якоба : как определить симптомы?

Болезнь Крейтцфельда-Якоба представлена в виде быстро развивающегося нарушения мозговой деятельности, для которого характерно снижение интеллектуальных функций.
Название заболевания обусловлено фамилиями немецких врачей – Ганса Герхарда Кройцфельда (в 1920 году впервые описал болезнь) и Альфонса Якоба (в 1921 году обеспечил значимый прогресс в исследовании заболевания).
Рассматриваемое явление относится к категории природных заболеваний, классифицирующихся как трансмиссивная нейродегенеративная прионная болезнь. Прионами принято называть белковые инфекционные частицы, провоцирующие патологию.
Заражение происходит путем трансформации здорового белка человеческого организма в подобный приону. Болезнь Крейтцфельда-Якоба считается наиболее распространенной среди других прионных недугов, а медики ее определяют как проявление губчатой энцефалопатии.
Причины болезни Крейтцфельдта-Якоба
- Существует несколько способов попадания прионов в организм человека.
- Наиболее распространенным вариантом считается прием зараженных лекарств или употребление инфицированной пищи.
- Заражение через не обеззараженные должным образом хирургические инструменты встречается намного реже, как и случаи инфицирования при пересадке органов.
Возникновение заболевания обусловлено нарушением метаболизма, для которого характерно излишнее накопление белка приона в нервной системе в измененной форме.
У здорового человека данный элемент также есть, у 1-2 людей из миллиона выявляется мутация, приводящая к губительным последствиям.
Заражение через еду
Допускается попадание в организм измененного белка посредством употребления зараженного коровьим бешенством мяса, другое его название – губчатый энцефалит. Статистика свидетельствует о том, что зараженное мясо стало причиной гибели 85 человек в Англии в 90-х годах.
С тех пор Евросоюз издал запрет об использовании в производстве костной и мясной муки туш животных, части которых потенциально опасные – глаза, позвоночник, мозги. Забитые на бойнях Европы животные, возраст которых превышает 30 месяцев, исследуются на наличие губчатого энцефалита.
Наследственное развитие
Наследственная причина считается менее распространенной. Мутантный ген передается от родителя. Наследственное заболевание отличается тем, что мутантный ген изначально экспрессирует неправильный белок, поскольку у организма хватает сил справляться самостоятельно, признаки патологии достаточно долго нельзя обнаружить.
Со временем в нейронах из-за нарушения функции белка откладываются прионные палочки. При достижении критического уровня функционирование нейронов прекращается, и начинают проявляться серьезные неврологические симптомы.
Во время активизации заболевания мозг больного становится, в буквальном смысле похож на губку. В среднем в течение 3-4 лет пациент погибает, поскольку лечения не существует. Зарегистрированы случаи, когда человек спустя несколько месяцев или недель после появления первых признаков умирал. Эксперты утверждают, что данная болезнь возникает практически всегда внезапно.
Случаи регулярной передачи патологии из поколения в поколения достаточно редкие.
Первые симптомы проявляются в возрасте 40-50 лет и до сих пор неизвестна причина столь позднего проявления мутирующего гена. Потомки могут получить недуг по наследству в 50% случаев.
Наибольший недостаток патологии заключается в том, что симптоматика возникает в том возрасте, когда у пациента есть дети или даже внуки.
Потомкам остается лишь провести генетический анализ и узнать, унаследовали они мутацию или нет.
При унаследовании мутации она гарантированно проявится в старости, при этом существует 50% вероятность рождения здорового потомства.
Следует упомянуть о наличии серьезной этической проблемы, поскольку далеко не у каждого человека хватает сил принять обреченность на смерть по причине данного недуга.
Формы
Существует четыре формы протекания данной трансмиссивной нейродегенеративной прионной болезни:
- Спонтанная форма считается классической, при этом наблюдается спонтанное проявление прионов, внешние предпосылки отсутствуют. Такое проявление характерно для людей пожилого возраста.
- Наследственная форма или, иными словами, генетическая.
- Ятрогенная форма является результатом инфицирования через зараженные медикаменты или недостаточным образом обеззараженные хирургические инструменты.
- Новая форма, причина которой представлена употреблением в пищу мяса инфицированных животных. Группа риска – люди в возрасте после пятидесяти и до тридцати.
Симптомы болезни Крейтцфельдта-Якоба
Отличительная особенность симптоматики болезни Крейтцфельда-Якоба заключается в постепенном ухудшении состояния пациенты. Актуально выделить три стадии.
- Симптомы продромального периода нельзя назвать специфичными, и характерны они для 30% пациентов. Первые проявления возникают за несколько недель или месяцев до первых признаков деменции. Для данного этапа характерно изменение поведения, потеря либидо, снижение веса, нарушение мышления, памяти, внимания, аппетита и сна, астения.
- Симптомы инициального периода – парестезии, неустойчивость, головокружение, головные боли и зрительные нарушения. Чаще всего развивается постепенный дебют, реже наблюдается острая и подострая форма. Иногда неврологические признаки могут свидетельствовать о будущей деменции, в частности при амиотрофических формах.
- Симптомы развернутого периода представлены прогрессирующим спастическим параличом конечностей. Данный признак сопровождают характерные движения, ригидность, тремор и экстрапирамидные знаки.
Деменция при болезни Крейтцфельдта-Якоба
Деменция при рассматриваемой патологии выражена прогрессирующим слабоумием приобретенного типа при наличии обширной неврологической симптоматики. Предположительно, именно гены влияют на специфические изменения, происходящие в нервной системе. Имеется в виду подострая спонгиформная энцефалопатия.
Источник: https://prodepressiju.ru/demenciya/bolezn-kreitcfeldta-yakoba.html
Болезнь Крейтцфельдта-Якоба
Болезнь Крейтцфельдта-Якоба — это тяжелое дегенеративное заболевание постоянно прогрессирующего характера, локализуется в базальных ганглиях, спинном или головном мозге. Другие названия патологии: псевдосклероз спастический, трансмиссивная спонгиоформная энцефалопатия, синдром кортико-стриоспинальной дегенерации, коровье бешенство.
Данное заболевание составляет 85% всех прионовых энцефалопатий, встречающихся у людей. Оно поражает одинаково интенсивно представителей всех рас, полов и возрастных групп. Незначительное преобладание частоты заболевания отмечается среди женщин на острове Новая Гвинея, с чем современные ученые часто связывают случаи каннибализма, традиционные для населения указанных территорий.
Попадая в человеческий организм, прион, который представляет собой инфекционную субмикроскопическую частицу, вызывающую дегенерацию головного мозга, задерживается на поверхности клетки и взаимодействует со здоровыми белками.
В результате их структура нарушается настолько, что внешне мозг человека напоминает губку. При этом естественные процессы в его больных клетках блокируются, что вызывает их быстрое разрушение.
Здоровые ткани, находящиеся рядом с очагом заболевания, воспаляются, и этот процесс приводит к постоянному усугублению состояния пациента.
Причины болезни Крейтцфельдта-Якоба
Основной причиной данного расстройства считается поедание мяса животных, болеющих спонгиозной энцефалопатией.
Кроме того, были отмечены случаи заражения пациентов в медицинских учреждениях через некачественно стерилизованные хирургические инструменты или через трансплантируемые ткани.
Описаны эпизоды заражения людей, подвергавшихся гормонотерапии, направленной на устранение дефицита гормона роста, когда лечение производилось с помощью экстрактов тканей, содержащих в себе данный гормон.
Кроме случаев инфицирования извне, существует наследственный фактор, вызывающий болезнь Крейтцфельдта—Якоба. Данная форма заболевания связана с мутацией в гене PRNP. Это нарушение вызывает появление в организме патологических прионов. Наследственное заболевание наблюдается в 10-15% семей, в которых когда-либо был поставлен диагноз любой формы данного расстройства.
Классификация болезни Крейтцфельдта—Якоба
Согласно классификации, заболевание Крейтцфельдта—Якоба подразделяется на 4 типа:
- Классический. В этой форме заболевания прионы поражают головной мозг без видимых причин. Подобный вид коровьего бешенства наблюдается чаще всего у людей старше 50 лет, встречается крайне редко — в 1-2 случаях на миллион. Проявляется инфекция потерей памяти, внезапными переменами настроения, снижением интереса к жизни. Впоследствии больной утрачивает способность обслуживать себя, теряет зрение и практически перестает говорить. На последних стадиях пациента беспокоят сильные галлюцинации.
- Наследственный. Течение стадий заболевания аналогично классическому типу, но причиной, вызвавшей болезнь, является генетическое наследование прионов.
- Ятрогенный. Данным типом заболевания пациент заражается при оперативном вмешательстве через лекарства, плохо стерилизованные инструменты, оболочки мозга, применяемые для закрытия ран. В 21 веке вероятность заражения болезнью Крейтцфельдта—Якоба вовремя операции практически нулевая.
- Новый вариант. Форма заболевания, при которой заражение происходит через мясную пищу, содержащую зараженные ткани. Известно о более чем ста случаях заболевания с 1995 года. Болезнь особенно часто поражает молодых людей, средний возраст которых равен 20 годам. Начальная стадия заболевания отмечается депрессией, потерей интереса к увлечениям и работе, замкнутостью и желанием одиночества. Далее наступает нарушение координации движений, отсутствие возможности самостоятельно есть и приводить себя в порядок. В отличие от классического типа, при данном варианте заболевания слабоумие наступает в последнюю очередь, и пациент осознает все происходящее с ним.
Симптомы болезни Крейтцфельдта–Якоба
Для раннего периода заболевания характерен такой симптом, как изменение личности, при котором человек проявляет беспокойство. Также для этой стадии характерны депрессии, бессонница, забывчивость и значительное ухудшение интеллектуальных способностей. Постепенно больной теряет зрение, что способствует утрате возможности адекватно обслуживать себя.
За этим следуют затруднения проглатывания пищи и воды, появления непроизвольных резких движений, которые невозможно предугадать. С прогрессированием заболевания состояние пациента ухудшается, что влечет за собой его впадение в кому.
Из этого состояния человек уже не выходит, а смерть наступает в результате развивающейся сердечной недостаточности, пневмонии и присоединения других инфекций.
У всех пациентов психиатрические симптомы присутствуют в начале заболевания, а позднее наступает фаза деменции. Поскольку болезнь неизлечима ее исход всегда только летальный.
Диагностика болезни Крейтцфельдта–Якоба
Способов постановки точного диагноза данной болезни в начале ее течения на сегодняшний день не существует. Специалисты могут предположить заболевание с помощью семейного анамнеза, неврологических тестов и некоторых диагностических процедур.
- В первую очередь больного направляют на электроэнцефалографию. Во время этой процедуры на голову обследуемого больного прикрепляются электроды, служащие для измерения электрической активности головного мозга. Нарушения в данной активности могут быть спровоцированы болезнью Крейтцфельдта—Якоба.
- Кроме электроэнцефалографии о развитии данного заболевания могут свидетельствовать и результаты МРТ. Это исследование основывается на применение магнитных полей высокой мощности для создания изображения тканей мозга на мониторе. Таким способом можно получить предельно качественную картинку серого и белого вещества, при осмотре которого можно выявить изменения, являющиеся признаками заболевания.
- Для постановки возможного диагноза проводится также анализ спинномозговой жидкости. Для этого производится люмбальная пункция и исследование полученного ее путем материала. Здесь признаком заболевания может быть наличие протеина, которого в здоровом состоянии быть не должно.
- Еще один этап обследования — биопсия миндалин, ткани которых способны накапливать в себе признаки прионной инфекции. Однако этот метод диагностики заболевания считается самым ненадежным из всех перечисленных.
- Наиболее надежным анализом при диагностировании коровьего бешенства врачи считают биопсию мозга — процедуру, во время которой у пациента забирают небольшое количество клеток с помощью специальной иглы. Это дает возможность осмотреть пораженную ткань вблизи и сделать самые точные выводы о том, насколько и какой инфекцией поражен головной мозг.
Лечение заболевания Крейтцфельдта – Якоба
В современной медицине не существует способов успешного излечения пациентов от данного заболевания. В последние десятилетия учеными были проведены тесты множества препаратов: антибиотиков, стероидов, иммуномодуляторов и антивирусных лекарств. Но все рассматриваемые методы показали полное отсутствие терапевтического эффекта.
Сегодня, работая с пациентами, страдающими болезнью Крейтцфельдта—Якоба, врачи могут проводить исключительно симптоматическое лечение, направленное на облегчение тяжести течения заболевания, но не на уничтожение прионов и оздоровление мозга. При подозрении на КБЯ, больному отменяют лекарственные препараты, негативно воздействующие на мнестические функции его организма.
Положительное влияние на состояние пациентов, а именно облегчение симптомов заболевания было выявлено при использовании следующих медикаментов: Брефелдина А и блокаторов кальциевых каналов.
Первые, разрушая аппарат Гольджи, создают препятствие для выработки PrPSc в структуре зараженных клеток.
Вторые останавливают работу NMDA-рецепторов, что становится причиной более длительного срока жизни инфицированных нейрональных культур.
Прогноз болезни Крейтцфельдта—Якоба
Практически во всех случаях данное заболевание приводит к смерти пациента в течение одного года.
Иногда летальных исход наступает ранее — через полгода после того, как был поставлен диагноз, но в особо редких случаях — приблизительно в 5% случаев — продолжительность заболевания может увеличиться до двух лет.
10% заболевших людей не проживают и нескольких недель, что вызвано индивидуальными особенностями их организмов.
Поскольку болезнь не передается воздушно-капельным путем, для медицинских работников и родственников, контактирующих с больными, ношение масок и других защитных средств не является обязательным условием.
Те сотрудники, которые проводят анализы биологических жидкостей и тканей пациентов, у которых подозревается болезнь Крейтцфельдта—Якоба, должны соблюдать правила безопасности и работать исключительно в перчатках. В особенности необходимо избегать прямого контакта слизистых оболочек с образцами зараженного материала.
Если такой крайне нежелательный контакт все же состоялся, необходимо в обязательном порядке произвести дезинфекцию 4% раствором гидроксида. Обработка проводится в течение 5-10 минут после контакта, после чего следует промывание слизистой проточной водой.
Обеззараживание инструментов проводится автоклавированием в течение часа при температуре 132°С либо часовой стерилизацией в 10% растворе гипохлорита натрия или 4% растворе гидроксида натрия. Стандартные способы стерилизации инструментов, такие как обработка формалином, в данном случае неэффективны.
Источник: https://www.mosmedportal.ru/illness/bolezn-kreyttsfeldta-yakoba/